| Page Index |
|
(医薬品と規制) |
|
|
|
(薬事法) |
|
|
|
(通知・事務連絡) |
|
|
|
(書面の電磁的保存・記録に関する法律) |
|
|
|
(関連法令等リンク) |
|
|
|
|
|
|
|
|
| |
|
|
|
医薬品、医薬部外品、化粧品及び医療機器は、『薬事法』という厳しい法律によって、原料から製造方法、ラベルに表示しなければならない内容や、広告の表現までキメ細かく規制されています。
薬事法によって必要な許認可・届出
化粧品製造販売業許可、化粧品製造業許可、
医薬品製造販売業許可、医薬品製造業許可、医薬品承認、
医薬部外品製造販売業許可、医薬部外品製造業許可、医薬部外品承認、
医療機器製造販売業許可、医療機器製造業許可、医療機器承認、医療機器修理業許可、
高度管理医療機器・特定保守管理医療機器販売業・賃貸業許可、管理医療機器販売業・賃貸業届出
|
|
|
|
|
|
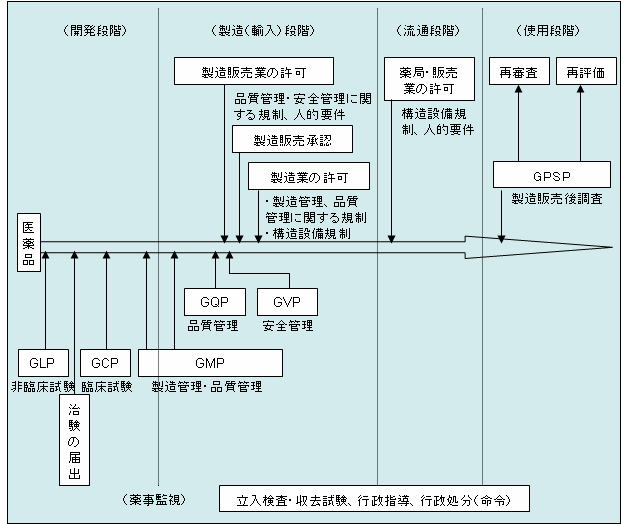 |
| 医薬品の承認審査等の現状について より |
|
|
|
|
|
|
|
|
|
|
|
ICH(International Conference on Harmonization) |
|
日・米・EUの3極間で、新医薬品の製造(輸入)承認に際して要求される資料を共通化することによって、医薬品開発の迅速化・効率化を目指そうという会議です。ICHの会議によって協議・合意決定された取り決め事項を「ICHガイドライン」と呼び、日米EUでの医薬品開発におけるガイドラインとしての役目を果たします。ICHは91年の発足以来、3極(日・米・EU)の持ち回りで2〜3年に1回のペースで開かれています。
ガイドラインがICHで合意(調和)に至ると、そのガイドラインを適用した医薬品開発や臨床試験、医薬品申請が各地域で可能となるよう、各国が法的な整備も含めた必要な措置を取ります。日本では、ICHで合意されたガイドラインは厚生労働省医薬食品局から通知されます。
|
|
ICHについての情報 |
|
|
|
|
|
ICH各会合の結果について |
|
ICH各会合の結果について
・2010年 6月 タリン
・2009年10月 セントルイス
・2009年 6月 横浜
・2008年11月 ブリュッセル
・2008年 6月 ポートランド
・2007年11月 横浜
・2007年 5月 ブリュッセル
・2006年10月 シカゴ
・2006年 6月 横浜
・2005年11月 シカゴ
・2005年 5月 ブリュッセル
・2004年11月 横浜
・2004年 6月 ワシントン |
|
| |
| (薬事法) |
|
|
|
薬事法は医薬品、医薬部外品、化粧品及び医療機器の品質、有効性及び安全性の確保のために必要な規制を行うとともに、医療上特に必要性が高い医薬品及び医療機器の研究開発の促進のために必要な措置を講ずることにより、保健衛生の向上を図ることを目的としています。
|
|
法律 |
|
|
|
|
|
施行令:政令 |
|
薬事法施行令 政令第11号、昭和36年1月26日 |
|
|
|
施行規則:省令 |
|
|
|
|
|
実施基準:省令 |
|
|
| |
| (通知・事務連絡) |
|
|
|
医薬品の安全性を確認するための毒性試験は、得られた結果は正確に解析・評価されるよう試験データの信頼性が確保されなくてはいけません。そのため、日本では医薬品の製造販売承認申請、再審査等に際して提出する各種毒性試験データは「医薬品の安全性に関する非臨床試験の実施の基準」、いわゆるGLPを遵守して実施されたものであることが義務づけられています。(2003年7月1日以降に実施される安全性薬理試験にも適用されます。)
|
|
|
|
|
|
|
|
|
|
|
|
旧GCP
治験を依頼しようとする者が依頼に際し遵守すべき基準及び治験が倫理的な配慮の下、科学的に適正に実施されるための基準である「医薬品の臨床試験の実施の基準」(1989年10月2日、薬発第874号、薬務局長通知。いわゆる旧GCP)が示され、1990年10月以降に治験実施計画書が作成された治験に適用されました。
ICH-GCP
1996年にICH-GCPが最終合意されました。 日本語訳
答申GCP
ICH-GCPを日本の医療環境・医療事情に合うようにしたものです。法的拘束力はありません。
医薬品の臨床試験の実施の基準(GCP)の内容 (中央薬事審議会答申) 1997年3月13日
新GCP
1997年4月1日より、治験のみならず市販後臨床試験においても被験者の人権保護、安全性の確保、臨床試験データの信頼性を確保するための基準である「医薬品の臨床試験の実施の基準」(1997年3月27日、厚生省令第28号。いわゆる新GCP)が施行されました。
改正GCP
2002年7月の薬事法改正のうちの一部が2003年に施行され、医師および医療機関が主体となって行う臨床研究のうち将来的な承認申請の意図を持って実施される治験(いわゆる医師主導の治験)が制度化され、医師および医療機関が未承認薬物の提供を受けて行う臨床試験や、既承認薬物を用いた適応外使用に関する臨床試験などを実施することができるようになり、2003年7月30日より施行されました(2003年6月12日、省令第106号、いわゆる改正GCP)。
医薬品と医療機器GCP 及びその運用の相違点 日本CRO協会
2008年改正GCPに関するQ&A 2009年3月12日、日本製薬工業協会
|
|
ヘルシンキ宣言 |
|
ヒトを対象とする医学研究の倫理的原則1964.6
ヘルシンキ宣言は、2008年10月に開催されたWMAソウル総会で大幅な修正が行われました。 |
|
|
|
|
|
省令(医薬品関係) |
|
| 医薬品の臨床試験の実施の基準に関する省令の一部を改正する省令 |
厚生省令第24号、
平成20年2月29日 |
医薬品の臨床試験の実施の基準に関する省令の一部を改正する省令
一部改正GCP省令 (治験審査委員会の質及び機能の向上) |
厚生省令第72号、
平成18年3月31日 |
医薬品の臨床試験の実施の基準に関する省令の一部を改正する省令
改正GCP省令 (薬事法改正にともなう用語の訂正) |
厚生省令第172号、
平成16年12月21日 |
医薬品の臨床試験の実施の基準に関する省令の一部を改正する省令
改正GCP省令 (医師主導治験の導入) |
厚生省令第106号、
平成15年6月12日 |
医薬品の臨床試験の実施の基準に関する省令
GCP省令(新GCP) |
厚生省令第28号、
平成9年3月27日 |
|
|
|
|
省令(医療機器関係) |
|
|
|
|
|
施行(医薬品関係) |
|
|
|
|
|
施行(医療機器関係) |
|
|
|
|
|
運用(医薬品関係) |
|
|
|
|
|
運用(医療機器関係) |
|
|
|
|
|
その他通知(医薬品関係) |
|
|
|
|
|
その他通知(医療機器関係) |
|
|
|
|
|
|
|
|
|
|
|
医療機器の承認申請に係る関連通知等 独立行政法人 医薬品医療機器総合機構
|
|
|
|
|
|
医薬品の市販後調査にあたり、その適正な実施と資料の信頼性の確保を目的として製薬企業のあるべき体制や実施規範が定められ、いわゆる新GPMSPとして1991年6月に行政通知として示され、1993年4月より適用されました。
さらに、市販後調査のより一層の充実強化を図るため1997 年3 月「GPMSP」として省令化され、4月1日から施行されました。(1997年3月10日、厚生省令第10号)
改正薬事法の施行に伴い、GPMSPは、製造販売後の安全管理の基準である「GVP」と製造販売後調査の実施の基準である「GPSP」に分けられ、GPSP省令は2005年4月1日から施行されました。
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
ICH E2B(R3) |
|
|
|
|
|
ICH E2B(R2)/M2
・E2B(R2) : 個別症例安全性報告を伝送するためのデータ項目
・M2 : 個別症例安全性報告を電送するためのメッセージ仕様
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
PIC/SのGMPガイドラインを活用する際の考え方(案)に関する意見募集について 2011年11月04日 厚生労働省医薬食品局監視指導・麻薬対策課 |
|
|
|
治験薬GMP |
|
|
|
|
|
医薬品・医薬部外品GMP |
|
|
|
|
|
|
|
|
|
|
|
|
|
医療機器の承認申請等に係る関連通知 リスト 独 医薬品医療機器総合機構 |
|
|
| (書面の電磁的保存・記録に関する法律) |
|
|
|
|
|
|
|
|
|
 |
|
|
|
(治験情報のIT化) |
|
|
|
|
|
|
|
|
|
法令 |
|
|
|
|
|
解説 |
|
ER/ESが求める要件
1.電磁的記録利用のための要件
・システムはバリデートされていること(CSV)
・セキュリティの保持の規則・手順の文書化されており適切に実施されていること
・監査証跡が自動的に記録されること
・バックアップ手順が文書化されており適切に実施されていること
・電磁的記録の内容を人が読める形式で出力ができること
・保存期間内において、真正性・見読性が確保された状態で電磁的記録が保持できること
2.電子署名利用のための要件
・電子署名の管理・運用に係る手順が文書化されており適切に実施されていること
・電子署名は各個人を特定できる唯一のものとし、他の誰にも再利用、再割り当てされないこと
・署名された電磁的記録には、署名者の氏名、署名が行われた日時、署名の意味を明示する項目が
含まれること
・電子署名は不正使用を防止するため対応する電磁的記録とリンクしていること
3.その他
・電磁的記録、電子署名の利用のために必要な責任者、組織、設備および教育訓練に関する事項を
規定しておくこと
|
ER/ES対応において整備すべき規定類
1.ポリシー
2.CSV
3.セキュリティ
4.バックアップ、リストア
5.電子署名
6.体制、監査証跡、電磁的記録の保存
7.教育訓練
|
ER/ES指針と、21 CFR Part 11で異なる点
「CAC 日本版ERESに関するFAQ」に詳しく解説されています。
|
|
|
|
|
参考になる解説、ガイドライン |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
条文およびガイドライン |
|
21 CFR Part 11 Final Rule(英文) FDA米食品医薬品庁
ガイドラインへのダイレクトリンクです。
|
|
21 CFR Part 11(英文) FDA米食品医薬品庁
制定の経緯、これまでのドラフト版、企業との議事録など公開されています。 |
|
|
|
適用範囲 |
|
この規則は、当局の規則で示されるすべての記録に関する要求下で、作成・変更・維持保管・検索、あるいは発信される電子形式の記録に適用されます。
当局の規則によって特に識別がない場合でも、連邦食品法、医薬品法、化粧品法、および公衆衛生サービス法において必要とされる機関への電子記録の提示にも適用されます。 |
|
|
|
規制の動向 |
|
・21CFR Part11(電子記録,電子署名に関する規則) 1997年8月20日付で発効
・21CFR Part11(業界向け)ガイダンス 2003年8月28日付で発効
(Part 11の解釈によって、予想外に膨大なコストが発生するなど複数の問題が発生したため、条文と関係ガイダンスを見直したもの) |
|
|
|
参考になる解説、サイトなど |
|
|
|
|
| (関連法令等リンク) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|










