| Page Index |
|
�i�����̋Ɩ��j�@���P�F�ʃy�[�W�փW�����v |
|
|
|
�i�����̃h�L�������g�j |
|
|
|
�i�@�K���j |
|
|
|
�i���a�n�w�j |
|
|
| �@ |
 �Տ������̂��Ƃ��悭�킩�� �Տ������̂��Ƃ��悭�킩�� |
  �Տ������E�����������T���N�v��2012�ĂɊւ���ӌ��̕�W�ɂ��� �@�����J���ȁA2012/03/01 �Տ������E�����������T���N�v��2012�ĂɊւ���ӌ��̕�W�ɂ��� �@�����J���ȁA2012/03/01 |
| �V���Ȏ����������T�J�N�v�� �@�����J���� |
| �S�������������R�J�N�v��ɌW�邱��܂ł̎��g�� �@�����J���� |
| �킪���̎����̌���Ɩ��_ �@�h�����w - ���_�����f�B�A |
| �@ |
|
|
|
�����̃v���Z�X�i�S�̑��j
�@�V���i�̊J���ɂ����ẮA���F�R���̂��߂̎�����肪�d�v�ł���A��Տ������y�їՏ������ɂ����ĐV���i�̕i���A�L�����y�ш��S�����������߂ɕK�v�Ȑ��т��K�v�ł��B�Տ������Ƃ��Ă͑�T���A��U���y�ё�V�������i���̓J�e�S���[�Ƃ��Ă̗Տ��I�����A�T���I�����A���ؓI�����A���ÓI�g�p�j���s���܂��B���Ɋe���ɂ�����Տ��������J�n�����ŁA��Տ��������邢�͐�s����Տ������̌��ʂɂ���܂̈��S�����\���Ɋm�ۂ����K�v������܂��B
�@�܂��A���F���邽�߂ɒ�o����鎑���͌����J����b�̒�߂��ɏ]���Ď��W����A���A�쐬���ꂽ���̂łȂ���Ȃ�Ȃ����Ƃ��@�ŋK�肳��Ă��܂��B�Տ������ɂ����ẮA�u���i�̗Տ������̎��{�̊�Ɋւ���ȗ߁v�iGCP�j���{�s����Ă��܂��B
�@ |
|
 |
|
�@ |
|
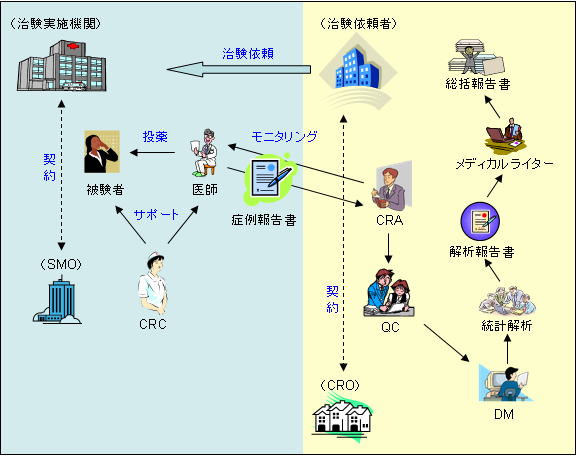
�������{�̃v���C���[ |
|
�����˗���
�@�����̔��āA�^�c�y�сi���́j�����ɐӔC���l�A��ЁA�����@�֖��͒c�́B
�@�ʏ�́A���[�J�[�܂��́A��Ë@�탁�[�J�[���w���܂��B
IRB�iInstitutional Review Board�j�F�����R���ψ���
�@�������{�@�ւ����������{����ۂɌ����J���Ȃɓ͂��o�������f�U�C����R�����钆���I�ȑg�D�ŁA�����̗ϗ����A���S���A�Ȋw�I�Ó�����R������ψ���ł��B
�������{�@��
�@���������{�����Ë@��
�b�q�n�iContract Research Organization�j�F���i�J���Ɩ�����@��
�@���i�̊J���ɂ����āA�����˗��ҁi�ʏ�A���[�J�[�j�̎����Ɋւ��Ɩ��̈ꕔ���s�E�x������l���͑g�D�E�c�́i�ʏ�́A��Ɓj�ł��B
�r�l�n�iSite Suport Organization�j�F�����{�ݎx���@��
�@���������{�����Ë@�ւɂ����鎡���Ɩ����s��x������g�D(�����{�ݎx���@��)�ł��B |
|
|
�@ |
|
�����̃v���Z�X�i�����˗��ґ��̃v���Z�X�Ɗe�����j |
|
���L�������i�Տ������v���Z�X�Ɗe�����j�ɋL�ڂ���Ă��܂��B
�@�@�f�[�^�}�l�[�W�����g�@������w��w�n�����ȁ@��×m�A2005�N
�@�@�@�@CBI���J�u���u�Տ��������@�_�v�@�Z�~�i�[���\���� |
|
|
�@ |
|
�����̃v���Z�X�i�������{�@�֑��̃v���Z�X�Ɗe�����j |
|
|
|
�@ |
|
�e�v���Z�X�̐��� |
|
CRC�̋L��
�@CRF�͈�t���쐬���܂��BCRC�͌���������̓]�L�̂�����܂��B
CRF�̉��
�@CRA�i�Տ��������j�^�[�j��CRF��������܂��B
�R�[�f�B���O
�@�a���╛��p�^�L�Q���ۖ����R�[�h�������Ƃł��B�ʏ�́A�f�[�^�}�l�W�����g���傪���{���܂��B
CRF�̃��r���[
�@��w�I�ϓ_����CRF�̐��������`�F�b�N���܂��B
�N�G���[�̔��s
�@CRF�f�[�^�̕s�����̔����ɂ���āA�������{���Ԃɑ��� ���s����Ɖ���ł��B
�@�N�G���̑����̓f�[�^�}�l�W�����g���傪���s���܂��B
�f�[�^�̓���
�@DM�i�f�[�^�}�l�[�W�����g����j��CRF���f�[�^�x�[�X�ɓ��͂��܂��B
�f�[�^�N���[�j���O
�@�f�[�^���̖�������������`�F�b�N���s���܂��B
SDV�iSourse Document Verification�j
�@�������ډ{���ɂ��ƍ����A�Ǘ���Ƃ̈�v�����m�F���A�����̓K�Ȏ��{�y�уf�[�^�̐M�������������܂��B
�Ǘጟ����
�@�W�߂�ꂽ�Ǘ���A���炩���ߌ��߂�ꂽ�v���g�R�[������ɉ�͏�ǂ̂悤�Ɏ�舵�����������A�����I�Ȍ�������錟����������܂��B�s���S�Ǘ�̎戵���A��͑ΏۂƂ��Ă̍̔ۂȂǂ����������ł��B��ɗՏ���𒆐S�Ƃ��Ă�����܂��B
�f�[�^�Œ�
�@CRF�̓o�^�ƑS���C�����I�������A������ł��Ȃ��悤�f�[�^�����b�N���܂��B
�L�|�I�[�v��
�@��d�ӌ������̏ꍇ�ɂ����āA�f�[�^�̌Œ��ɖ�܊��t���ʁi�R�[�h�j�𖾂炩�ɂ��邱�Ƃł��B
��܊��t���ʁi�R�[�h�j�́A�������{�҂���Ɨ��������t�ӔC�҂��ۊǁE�Ǘ����܂��B |
|
|
�@ |
|
�����ɂ�����u�t�F�[�Y�v |
|
��T�������i�ł���\�I�Ȏ����F�Տ������j
�@�@��T���́A����������߂ăq�g�ɓ��^���邱�Ƃ���J�n�����B�ł���\�I�Ȏ����̎�ނƂ��ẮA�Տ�����
�@����������B�Տ������͒ʏ��T���Ɠ���ł��邪�A��A�̊J���̉ߒ��̒��ő��̑��ōs���邱�Ƃ���
�@��B��T���̖ړI�ɂ͒ʏ�@�ȉ��̈���邢�͑g�������܂܂��B
�@�@�@�@ �����̈��S���y�єE�e���̕]��
�@�@�@�A ���Ԃ̌���
�@�@�@�B ��͊w�I�ȕ]��
�@�@�@�C �����̖���]��
�@�Q�l�Ƃ��ׂ������Ƃ��āu���i�̗Տ����Ԏ����ɂ��āv�i2001�N6��1���A���R����796���j����������B
��U�������i�ł���\�I�Ȏ����F�T���I�����j
�@��U���́A�ʏ튳�҂ɂ����Ď��Ì��ʂ�T�����邽�߂̎������J�n����i�K�ł���B
�@�T�^�I�ȑ�U���́A�@���m�ɒ�`���ꂽ��ɏ]���đI������A���̏�Ԃ��ώ@����Ă��銳�ҌQ��ΏۂƂ���
�@�s������̂ŁA��\�I�Ȏ����Ƃ��ĒT���I��������������B���̑��̏d�v�ȖړI�͑�V���ŗp����p�@�E�p��
�@�����肷�邱�Ƃł���B
�@�@��U���O��
�@�@�@�E�����߂Ċ��҂Ɏ��݂�i�K
�@�@�@�E�Տ��p�ʂ͈̔́A�K�������͈̔͂�T��
�@�@��U�����
�@�@�@�E�K���ƂȂ銳�ҁi�����j��Ώ�
�@�@�@�E���K�p�@�p�ʂ�ݒ�
��V�������i�ł���\�I�Ȏ����F���ؓI�����j
�@��V���͎��Ì��ʂ̌�����v�ȖړI�Ƃ��鎎���ł���B��V���̎�v�Ȏ����́A�Ӑ}�����K���Ⓤ�^�����
�@���ҌQ�ɂ����Ă��̖�܂����S�ŗL���ł���Ƃ�����U���܂łɒ~�ς��ꂽ�\���I�ȍ����������邽�߂�
�@�f�U�C�������B���̎����͐������F�̂��߂̓K�ȍ����ƂȂ�f�[�^�邱�Ƃ��Ӑ}���Ă���B
��W�������i���l�Ȏ����F���ÓI�g�p�j
�@��W���Ŏ��{����鎎���́A���i�̏��F��ɊJ�n����A���F���ꂽ�K���Ɋ֘A������̂ł���B
�@�s�̌�̕���p�����p�x������g�p���ђ����A���ʂȊ��҂�ΏۂƂ������ʒ����A�s�̌�Տ����������A
�@����ɊY������B�킪���ł́A��W���ƌď̂�����APMS�i�����̔��㒲���j�ƌĂ�Ă���B |
|
|
���{�̖s���@���{����H�Ƌ���A2007�N3�� ��� |
|
�@ |
|
�����ɂ�����u�t�F�[�Y�v �F �R����܂̏ꍇ |
|
�R������ᇖ�̗Տ��]�����@�Ɋւ���K�C�h���C��
��T������
�@��T�������͔�Տ��������т���Ɏ���������߂ăq�g�ɓ��^����i�K�ł���B��Տ������Ŋώ@���ꂽ���ۂɊ�Â��A�p�ʂɈˑ�����������̈��S������������̂���ȖړI�ŁA������̓��^�o�H�A���^�X�P�W���[���A�ő�ϗʁiMTD�j���͍ő勖�e�ʁiMAD�j�A�p�ʐ����Ő��iDLT�j�A���ԂƓŐ��̊֘A���A��U�������ɂ����鐄���p�ʂ����߂�B�i��ʓI�Ȗ�܂̑�T�������́A���퐬�l�j�q�{�����e�B�A��ΏۂƂ��čs�����A�j�Ő��������R������ᇖ�̑�T�������ł́A���N�Ȑl�ł͂Ȃ��A���҂�ΏۂƂ��ׂ��ł���B�܂��A��ʓI�ɔF�߂�ꂽ�W���I���Ö@�ɂ���ĉ�����Ǐ�ɘa��������\���̂��邪�҂�ΏۂƂ��ׂ��ł͂Ȃ��B
��U������
�@��U�������́A����̊���ɑ���L�����A���S����]�����邽�߂Ɏ��{����鎎���ŁA�ΏۂƂ������ɂ����鎡����̗Տ��I�Ӌ`�̂��鎡�Ì���(�ʏ�A��ᇏk������)�A�y�ш��S����]������B
��V������
�@��V�������́A���D�ꂽ�W���I���Ö@���m�����邽�߂ɍs����Տ������ł���B��U�������ɂ����Ĉ��S���Ǝ�ᇏk�����ʁA���͉��炩�̃����b�g�i�Ǐ�ɘa���ʓ��j���m�F���ꂽ�V�K�R������ᇖ�̒P�Ɩ��͕��p�Ö@�ƓK�ȑΏƌQ�Ƃ̔�r�����ł���B���Ґ�����������i�זE�x���A�݊��A�咰���A�������j��ΏۂƂ����R������ᇖ�ł́A���ꂼ��̊���ɂ��ĉ������ʂ𒆐S�ɕ]�������V�������̐��т����F�\�����ɒ�o���邱�Ƃ�K�{�Ƃ���B |
|
|
(����17 �N11 ��01 �� ��H�R������1101001 ��)��蔲�� |
|
�@ |
|
�����ƗՏ������̈Ⴂ |
|
����
�@�Տ������̂����A�V�������i���Ë@��̐����̔����F�������J���Ȃ��瓾�邽�߂Ɏ��{���鎎����\���܂��B
�@
�Տ�����
�@�V��̊J���̖ړI�Ɍ���Ȃ��A�l��ΏۂƂ������Â����˂������������A�ꕔ�������܂܂�܂��B
�@
�Տ�����
�@�Տ����������łȂ��A�Ǘ�⒲�����܂߂�������\���܂��B
��w�����@ |
|
|
�@ |
|
|
|
CRA�i�Տ��������j�^�[�j |
|
�@��Ë@�ւƂ̌_��A������̌�t�A�Տ������̎��{�A�Տ��f�[�^�̉���Ȃǂ��s���܂��B |
|
|
|
�@ |
|
DM�i�f�[�^�}�l�[�W���[�j |
|
�@�f�[�^�̓��́A�`�F�b�N�A�C���A�Ǘ���ʂ��ĉ�����ꂽ�Ǘ���iCRF�j�̃f�[�^���f�[�^�x�[�X�����܂��B
��̓I�ɂ́A�v���g�R���̍쐬�A�Ǘ���v�A�f�[�^�x�[�X�\�z�E�Ǘ��A���҃f�[�^�̓o�^�A�f�[�^���́E�����A�o���f�[�V�����A��͌��ʋy�ѕ��̃��r���[�Ȃǂ��s���܂��B
�@���ۂɂ́AIT�Ɋ֘A����Ɩ��ɏ]������l�ƁA�^�p�Ɋ֘A����Ɩ��ɏ]������l�́A������čs���邱�Ƃ������B |
|
|
|
�@ |
|
�������v�� |
|
�@�������v�w�̎�@��p���āA�����̌��ʂ���͂��A������̗L�����y�ш��S�����A���v�w�I�ɐ����i���j���܂��B
��̓I�ɂ́A�v���g�R���̍쐬�A�Ǘ���v�A���v��͌v�揑�̍쐬�A��̓v���O�����̍쐬�A���v��͕��̍쐬�A�o���f�[�V�����Ȃǂ��s���܂��B
�������v�Ƃ̐��͌��݁A400�l���x�Ɛ��肳��Ă��܂��B |
|
|
|
�@ |
|
CRC�i�����R�[�f�B�l�[�^�[�j |
|
�@CRC�͎����ӔC��t�̎w���E�ē̂��ƁA���I���ꂩ�玡���ӔC(���S)��t�̋Ɩ��ɋ��͂��܂��B
�Ɩ��Ƃ��ẮA���҂ɑ������̐������s������A�����̃X�P�W���[���Ǘ��A�e��f�[�^�̎��W��Ǘ��ȂǍs���܂��B
CRC�̐��͌��݁A5,000�l���x�Ɛ��肳��Ă��܂��B |
|
|
|
�@ |
|
���f�B�J�����C�^�[ |
|
�Տ������v��̗��āE�����A�Տ������̑������Ȃǂ̏��F�\���������쐬���܂��B
���f�B�J�����C�^�[�ɂ͉u�w�A�������v�w����@�_�̒m�������߂��܂��B |
| �@ |
| �����̌���Ɖۑ� |
|
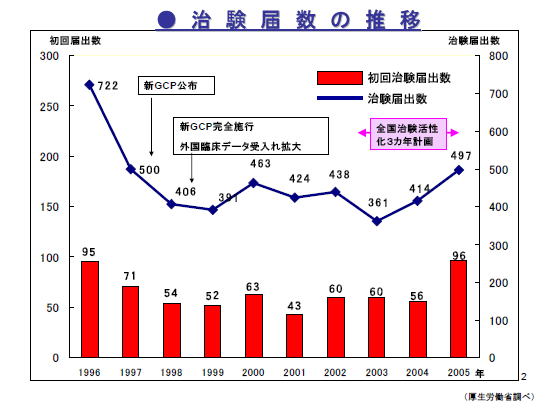
�@���i�͈��S���Ȃǂׂ�Տ������Ȃǂ�����A�V���������̔��������̎��p���܂łP�O�N�ȏ�B���p���܂ŕ��ςT�N���x�Ƃ����Ă��܂��B
�@����ɓ��{�̎���Ƃ��č����̎������Ԃ��č��ɔ�ׂP�N�ȏ㒷���B���F�R�����܂߂�Q�N���Ă��܂��B���S������A���Ԃ��Z�����Ƃ����ׂĂ����Ƃ͂����܂��A���{�̏��F���x�ɐV���܂�ɂ����ő�̌���������Ƃ����w�E�͑�������܂��B
�@�܂��A���{�͍��̏��F���x�����łȂ��A���҂ւ̌��ʂ��Ȋw�I�ɒ��ׂ�Տ��������ア�Ƃ����Ă��܂��B
�@�Ĉ�w�G���j���[�C���O�����h�E�W���[�i���E�I�u�E���f�B�V�����P�X�X�P�N����P�O�N�ԂɌf�ڂ��ꂽ�����Ȋw����̍��ʘ_���V�F�A�ׂ��Ƃ���A��b�Ȋw�ł͂S�ʂ��������{�͗Տ������ł͂P�S�ʂł����B�����ɎQ�����銳�҂��W�܂�ɂ����Ȃǂ̗��R������܂��B
�@���̂��߁A�Q�O�O�O�`�Q�O�O�S�N�ɉ��ĂȂǂŏ��F���ꂽ�V��P�U�T��̂����u�l���̎O�����{�Ŏg�����A���҂̕s���v�ɂȂ��Ă��錻����܂��B |
|
 |
|
�����J���Ȏ����@�u�S�������������R�J�N�v��ɌW�邱��܂ł̎��g�݁v�@��� |
|
�@ |
|
�������̌������Ɋւ�����@�����J���ȁA2011/07/05
�@�@ |
�����Ƃɑ��鎡���̌���A���P�[�g���������@���{����H�Ƌ���A2008�N3��27��
�@���i�]���ψ���Տ��]������ł́A2003�N�x���Տ��]�����������Ђ�ΏۂɎ����̎��Ԃɂ��Čp���I�ɒ������Ă���܂��B�����̌���ɂ��ĊW�҂��������L���A����̎������i�ɖ𗧂ĂĂ����������߁A2007�N�x�������ʂ̈ꕔ���f�ڂ��Ă��܂��B
�@ |
�g����x��h�ȓ��{�̈��i�@Biotechnology Japan
�@�@�g����x��h�ȓ��{�̈��i�i�O�ҁj�@2006�N12��12��
�@�@�@�@�Ȃ������Ŏ����͐i�܂Ȃ��̂��H
�@�@�g����x��h�ȓ��{�̈��i�i��ҁj�@2006�N12��20��
�@�@�@�@�R�������͕č���10����1�ȉ���Ƃ̎������k�ɂ��Ή��ł���
�@ |
�i�܂ʈ�t�哱�����@�ǔ��V���A2005�N9��17��
�@���̖����F��g�p��茟����c���A�����Ђɂ�鎡��������ȏꍇ�A��t�哱��������������Ƃ��Ă��܂��B���{��t��͂Q�O�O�R�N�A���̕⏕������Ɂu�������i�Z���^�[�v��ݗ��A��t�哱������i�߂Ă��܂��B�w����t�哱������v�]���ꂽ��͖�Q�T�O��ނɏ��܂��B�����A���ۂɎ������v�悳�ꂽ�̂͂P�O�܂ŁA���{����Ă���̂͂Q�܂ɉ߂��܂���B
�@ |
���������������Ȃ��u���������v�̗��R�@���o���f�B�J�� �I�����C���A2006�N7��5��
�@���J�Ȉ㐭�nj����J���V���ۂ́A���{�̎������u�x���A�����A���������v���Ƃ̗��R�Ƃ��āA�i1�j�팱�ҁi���ҁj�̃C���Z���e�B�u���Ⴂ�A�i2�j���{�����҂̃C���Z���e�B�u���Ⴂ�A�i3�j�����̎��{�̐����ア�A�i4�j�����𒅎��Ɏ��{�����t�E��Ë@�ւ��s�����Ă���|�|�������܂����B
�@ |
�u�����̋��v���~�܂�Ȃ��@���o���f�B�J�� �I�����C���A2006�N6��28��
�@98�N�Ȍ�A�����͂��o���͔N��400�����x�ɂƂǂ܂��Ă���A�ȑO�ɔ�ׂ���Ȃ茸�����Ă��܂��B�������ł��Ȃ��A���邢�͎����̊J�n���x���Ȃ�A���F���x���̂����R�ł��B
�@ |
|
|
�@ |
|
|
|
|
|
�@ |
| �i�����̃h�L�������g�j |
|
|
|
�v���g�R���́A���������{�ɂ���ɂ������āA�������{�ҁi���������{�����Ë@�ցj�y�ю����˗��ҁi���[�J�[�j�����炵�Ȃ���Ȃ�Ȃ����̎����Ɋւ���v��������S�Ėԗ��L�ڂ������{�v�揑�ł��B�����͂��̃v���g�R�������炵�čs���܂��B
�@�@�v���g�R���ɕK�v�ȍ���
�@�@�@�E�ړI
�@�@�@�E�w�i�ƍ���
�@�@�@�E������/�������i���
�@�@�@�E�f�f��ƕa���E�a�^����
�@�@�@�E�K�i�K��?�o�^�E���t
�@�@�@�E���Ìv��
�@�@�@�E�L�Q���ۂ̕]���E��
�@�@�@�E�ώ@�E�����E���ڂƃX�P�W���[��
�@�@�@�E�ڕW�Ǘᐔ�Ǝ�������
�@�@�@�E�G���h�|�C���g�̒�`
�@�@�@�E���v�w�I�l�@
�@�@�@�E�Ǘ���̋L���ƒ�o
�@�@�@�E���j�^�����O
�@�@�@�E�ϗ��I����
�@�@�@�E�����̔�p���S
�@�@�@�E�v���g�R���̉���
�@�@�@�E�����̏I���Ƒ������~
�@�@�@�E�L�^�̕ۑ�
�@�@�@�E�������ʂ̋A���ƌ��ʂ̌��\
�@�@�@�E�����g�D?����
�@ |
|
|
|
�@ |
|
|
|
�������{�v�揑�ŋK�肳��Ă���팱�҂̑S�Ă̏����L�^���邽�߂̕��ł��B
CRF�͏����₷���v����Ă���K�v������܂����A���̌`���ɂ���Ď��̂Q�ɕ��ނ���܂��B
���q�^�iBook�j�Ǘ��
�@�Ǘ�P�ʂň���쐬���܂��B�͓��^�I����ɂ܂Ƃ߂čs���܂��B
�@�ώ@�E������ƂɋL�ڂ���Ă���̂ňꗗ��������Ƃ��������b�g������܂��B
�����^�iVisit�j�Ǘ��
�@�L�������ł��Ƃɉ���\�ȃ��[�t�^�̏Ǘ��.�����B�͕������ɍs���܂��B
�@�{�����[�����傫���Ȃ�Ƃ������_������܂����A������^�C�����ɍs����̂ŁA�L�ڃ~�X�A�v���g�R����E�Ȃǃf�[�^�̕s��𑁊��ɔ����ł��郁���b�g������܂��B
�@ |
|
|
|
�@ |
|
|
|
�@�����Ɩ���N�����{���Ă��K���ώ��ɐ��s�ł���悤�Ɋ�{�I�ȋƖ��菇��̌n�I�ɂ܂Ƃ߂��菇���ł��B
�VGCP�ɂ����ẮA�����Ɍg���A���[�J�[�i�����˗��ҁj�ACRO�A��Ë@�ցi�������{�{�݁j�́ASOP���쐬���A����Ɋ�Â���Ƃ��s�����Ƃ��`���Â����Ă��܂��B
�@SOP�̓��e�ɂ��ẮA�e�ГƎ��̎菇�ŗǂ��Ƃ���Ă��܂��B
|
|
�i�������{�@�ւ̃T���v���j |
|
|
|
�@ |
|
|
|
�L�ڗ�
�@�@�@�E�T�v�i�Ɩ��͈́j
�@�@�@�E���{�̐��i�ӔC�ҁA�S���ҁj
�@�@�@�E�Ɩ��菇
�@�@�@�E�����܂ł̎菇
�@�@�@�E���p����V�X�e��
�@�@�@�E���^�f�[�^
�@�@�@�E�V�X�e���o���f�[�V�����̃|���V�[
�@�@�@�E�p�ӂ���}�j���A����
�@�@�@�E�f�[�^���̗͂���
�@�@�@�E�R�[�f�B���O
�@�@�@�E�L�Q���ۂ̕�
�@�@�@�E�I����
�@�@�@�E�����̕ۊǕ��@ |
|
�@ |
|
|
|
|
|
�@ |
|
|
|
�����̏I����A�����̖ړI�A���@�y�ѐ��ѓ����܂Ƃ߂������Ɋւ�����ł��B���̍\���Ɠ��e�ɂ��Ă̓K�C�h���C��������Ă���̂ł���ɉ����č쐬����K�v������܂��B�����������͗ʓI�ɂ��c��Ȃ��̂ɂȂ�܂��B
|
|
|
| �@ |
| �i�@�K���j |
|
|
|
�Տ������Ɋւ���K�C�h���C�� |
|
������ |
|
|
|
�����I�v�� |
|
|
|
GCP�i���i�̗Տ������̎��{�̊�j |
|
|
|
�Տ����� |
|
|
|
�Տ��]�� |
|
|
| �@ |
|
|
|
�����V�X�e���̋K���ɑ���Minimum Requirment�Ƃ��āA���̂悤�ȑΉ������߂��܂��B |
|
�i�P�j�p�b�P�[�W�\�t�g�E�F�A�̏ꍇ�iConfigurable Softawre Pakages�j
�@�EValidation Plan�iVP�j���쐬���邱��
�@�ETraceablity Matrix(TM)���쐬���邱��
�@�EUser Requirements Specifications�iURS�j���쐬���邱��
�@�EFunctional specifications�iFS�j���쐬���邱��
�@�ETechnical specification�iTS�j���쐬���邱��
�@�EInstallationQualification�iIQ�j���s������
�@�EOperation Qualification�iOQ�j���s������
�@�EPerformance Qualification�iPQ�j���s������
�@�EValidation Report�iVR�j���쐬���邱��
�@�E�h�L�������g�A�v���O�����A�f�[�^�͒���I�Ƀo�b�N�A�b�v���邱��
�@�E�v���O���}�[�ɑ��ċ���E�P�����s������
�@�E�V�X�e�����^�p����SOP���쐬���邱��
�@ |
�i�Q�j�f�[�^�}�l�[�W�����g�ɂ����郍�W�J���`�F�b�N�̏ꍇ�iCustom Software�j
�@�EValidation Plan�iVP�j���쐬���邱��
�@�E�v���O�����d�l�����쐬���邱��
�@�E�v���O�����e�X�g�̓e�X�g�d�l�����쐬�����{���邱��
�@�EValidation Report�iVR�j���쐬���邱��
�@�E�h�L�������g�A�v���O�����A�f�[�^�̊č��ؐՂ��L�^���邱��
�@�E�h�L�������g�A�v���O�����A�f�[�^�͌����̂�����݂̂̂��A�N�Z�X�ł���悤�Ȏd�|����������
�@�E�h�L�������g�A�v���O�����A�f�[�^�͒���I�Ƀo�b�N�A�b�v���邱��
�@�E�v���O���}�[�ɑ��ċ���E�P�����s������
�@ |
�i�R�j���v��͂ɂ�����r�`�r�v���O���~���O�̏ꍇ�iCustom Software�j
�@�EValidation Plan�iVP�j���쐬���邱��
�@�E�v���O�����d�l�����쐬���邱�ƁB�����̃h�L�������g������ꍇ�͑�p���Ă��ǂ�
�@�E�v���O�����e�X�g�̓e�X�g�d�l�����쐬�����{���邱��
�@�EValidation Report�iVR�j���쐬���邱��
�@�E�h�L�������g�A�v���O�����A�f�[�^�̊č��ؐՂ��L�^���邱��
�@�E�h�L�������g�A�v���O�����A�f�[�^�͌����̂�����݂̂̂��A�N�Z�X�ł���悤�Ȏd�|����������
�@�E�h�L�������g�A�v���O�����A�f�[�^�͒���I�Ƀo�b�N�A�b�v���邱��
�@�E�v���O���}�[�ɑ��ċ���E�P�����s������
�@ |
�i�S�j�N�G���Ȃǔ��s���ăf�[�^�Q�Ƃ���ꍇ�iCustom Software�j
�@�E�N�G���[�ƌ��ʂ͕R�t���ĊǗ����邱��
�@�E�v���O���}�[�ɑ��ċ���E�P�����s������ |
|
|
�@ |
| �i���a�n�w�j |
|
|
|
|
|
�@ |
|
|
|
|
|
�@ |
|
|
|
���i�J���̌���ƍ��ۋ��������Ɋւ����{�I�l�����@2007�N12��
�@�v�ʐ����Z�~�i�[
�@, |
���ۋ��������Ɋւ����{�I�l�����ɂ����@��H�R������0928010���A2007�N9��28��
�@�����J���Ȉ��H�i�ǐR���Ǘ��ے��ʒm
�@, |
���{���܂ލ��ۋ����������{���@���j���[�XNo.21�@���Y�Ɛ������A2006�N11��8��
�@���ۋ��������Ƃ��Ď��{����Ă���v���g�R���S�P�R���̂����A���{�ł̎��{���܂ނ��̂͋͂��U���ɉ߂��Ȃ����Ƃ��A���Y�Ɛ������̒����ŕ�����܂����B��P�ʂ������č��̂Q�U�S���ɔ�ׂ�Ɨ]��ɂ����Ȃ��B
�@�@���{���܂ލ��ۋ����������{�@���Y�Ɛ�������C���������c��
�@�@�@�|�č�������w�}���َ����o�^���p���������|
�@ |
|
|
�@ |
|
|
|
|
|
�@ |
|
|
|
|
|
�@ |
|
|
|
|
|
�@ |
|
|
|
|
|
�@ |
|
|
|
|










