| Page Index |
|
(非臨床試験の業務) ※1:別ページへジャンプ |
|
|
|
(GLP試験において保存すべき資料) |
|
|
|
(法規制) |
|
|
|
(情報BOX) |
|
|
|
|
| (非臨床試験の業務) |
|
|
|
新医薬品の開発においては、承認審査のための資料作りが重要であり、非臨床試験及び臨床試験において新医薬品の品質、有効性及び安全性を示すために必要な成績を得る必要があります。
非臨床試験としては、理化学的試験、薬理・薬物動態・毒性に関する試験があります。非臨床試験は、以前は前臨床試験とも呼ばれていましたが、臨床試験開始後にも行われることから非臨床試験としています。非臨床試験の結果,有効性が期待でき,安全性にも問題がないと考えられた場合にヒトで行うのが臨床試験です。
なお、承認を受けるために提出される資料は厚生労働大臣の定める基準に従って収集され、かつ、作成されたものであることが薬事法で規定されています。非臨床試験においては、「医薬品の安全性に関する非臨床試験の実施の基準に関する省令」(GLP)が施行されています。 |
|
|
|
|
|
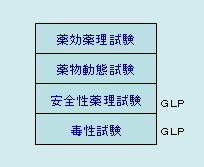
医薬品の承認申請に必要な非臨床試験は次の4種類が義務づけられ、それらを経て安全性・有効性が認められたものだけが次の臨床試験に進むことができます。 |
|
 |
|
薬効薬理試験
くすりの候補である化合物が、どの程度の効き目をもっているのか、効き目の持続時間はどれくらいか、どのような副作用が生じる可能性があるのかなど、生体内での実際的なくすりの効き目を動物を使って試験します。
薬物動態試験
くすりは体のなかで、吸収(Absorption)、分布(Distribution)、代謝(Metabolism)、排泄(Excretion)という段階をへて、尿などとして体外に排出されます。しかし、くすりの種類や性質によって、体内での吸収のされ方などに違いがあるため、血中濃度や臓器への分布を調べ、候補化合物が実際に体内でどのような状態を示すのかを試験します。
略してADME(薬物動態)試験と呼ばれます。
非臨床薬物動態試験ガイドラインについて 医薬審第496号、平成10年6月26日
反復投与組織分布試験ガイダンス 薬審442、平成8年7月2日
安全性薬理試験(一般薬理試験)
薬物の薬理作用または副作用の観察を目的として、ヒトでの安全性を予測するために行われる試験です。
大量投与されたときなどに主な生理機能に対して医薬品としてどのような望ましくない作用があるのかを調べます。
安全性薬理試験は毒性試験との関連が深いことから、GLP試験として取り扱われます。
安全性薬理試験ガイドラインについて 医薬審発第902号、平成13年6月21日
毒性試験
どのようなくすりも、量が多すぎたり、使い方によっては危険なものとなります。試験によって安全な領域を確かめたり、毒性(副作用)の種類などを調べます。
新有効成分医薬品の場合、毒性試験は「単回投与毒性試験」、「反復投与毒性試験」、「遺伝毒性」、「生殖発生毒性試験」を実施することが前提で、必要に応じ、「局所刺激試験」、「がん原性試験」(臨床で6ヶ月以上投与される薬剤)や「依存性試験」(向精神薬など)を実施することになります。
医薬品の免疫毒性試験に関するガイドラインについて 薬食審査発第0418001号、平成18年4月18日
医薬品の遺伝毒性試験に関するガイドラインについて 医薬審第1604号、平成11年11月1日
医薬品のがん原性試験に関するガイドラインについて 医薬審第1607号、平成11年11月1日
※医薬品の安全性を確認するための毒性試験は、得られた結果は正確に解析・評価されるよう試験データの信頼性が確保されなくてはいけません。そのため、日本では医薬品の製造販売承認申請、再審査等に際して提出する各種毒性試験データは「医薬品の安全性に関する非臨床試験の実施の基準」、いわゆるGLPを遵守して実施されたものであることが義務づけられています。(2003年7月1日以降に実施される安全性薬理試験にも適用されます。)
|
|
|
|
|
参考になるサイト |
|
|
|
|
| (GLP試験において保存すべき資料) |
|
|
|
医薬品の安全性に関する非臨床試験の実施の基準に関する省令 第七条六において、「試験中及びその終了時に以下の試験関係資料が保存する施設に保存されていること」を求めています。
試験計画書
標本
生データ
その他の記録文書
最終報告書
これらの変更又は訂正に係る文書 |
|
|
|
|
|
運営管理者は、次に掲げる事項に関する実施方法及び手順を記載した標準操作手順書を作成しなければならない。 医薬品の安全性に関する非臨床試験の実施の基準に関する省令 第十一条)
一 被験物質及び対照物質の管理
二 施設設備又は機器の保守点検及び修理
三 動物飼育施設の整備
四 実験動物の飼育及び管理
五 実験動物の一般症状等の観察
六 試験の操作、測定、検査及び分析
七 ひん死の動物及び動物の死体の取扱い
八 動物の剖検及び死後解剖検査
九 標本の採取及び識別
十 病理組織学的検査
十一 生データの管理
十二 信頼性保証部門が行う業務
十三 試験従事者の健康管理
十四 その他必要な事項 |
|
|
|
|
|
試験責任者は、試験ごとに、次に掲げる事項を記載した試験計画書を作成し、運営管理者(試験が委託された場合にあっては、試験委託者及び運営管理者)の承認を受けなければならない。 医薬品の安全性に関する非臨床試験の実施の基準に関する省令 第十五条)
一 表題と試験目的
二 試験施設の名称及び所在地
三 試験が委託された場合にあっては、試験委託者の氏名及び住所
(法人にあってはその名称及び主たる事務所の所在地)
四 試験責任者の氏名
五 被験物質及び対照物質に関する事項
六 試験系に関する事項
七 試験の実施方法に関する事項
八 生データの解析に使用する統計学的方法に関する事項
九 その他保存される記録及び資料に関する事項
十 運営管理者及び試験責任者の署名又は記名なつ印及びその日付
十一 その他試験の計画のために必要な事項 |
|
|
|
|
|
試験責任者は、試験ごとに、次に掲げる事項を記載した最終報告書を作成しなければならない。 医薬品の安全性に関する非臨床試験の実施の基準に関する省令 第十七条)
一 表題と試験目的
二 試験施設の名称及び所在地
三 試験の開始及び終了の日
四 試験責任者その他の試験に従事した者の氏名
五 被験物質及び対照物質に関する事項
六 試験系に関する事項
七 予見することができなかった試験の信頼性に影響を及ぼす疑いのある事態及び試験計画書に従わなかったこと。
八 試験の実施方法に関する事項
九 生データの解析に使用された統計学的方法に関する事項
十 試験成績及びその考察並びにこれらの要約
十一 生データ及び標本の保存場所
十二 試験責任者の署名又は記名なつ印及びその日付
十三 第八条第一項第八号の規定により信頼性保証部門責任者が作成し、署名又は記名なつ印した文書
十四 その他必要な事項 |
|
|
| (法規制) |
|
|
|
GLPシステムの規制に対するMinimum Requirmentとして、次のような対応が求められます。 |
|
(1)パッケージソフトウェアの場合(Configurable Softawre Pakages)
・Validation Plan(VP)を作成すること
・Traceablity Matrix(TM)を作成すること
・User Requirements Specifications(URS)を作成すること
・Functional specifications(FS)を作成すること
・Technical specification(TS)を作成すること
・InstallationQualification(IQ)を行うこと
・Operation Qualification(OQ)を行うこと
・Performance Qualification(PQ)を行うこと
・Validation Report(VR)を作成すること
・ドキュメント、プログラム、データは定期的にバックアップすること
・プログラマーに対して教育・訓練を行うこと
・システムを運用するSOPを作成すること |
|
|
|
|
参考になるサイト |
|
|
|
|
| (情報BOX) |
|
|
|
|










