| Page Index |
|
|
|
(別ページへジャンプ) |
|
|
| |
|
|
|
安全性情報とは、臨床試験や市販後において、被験者あるいは患者に起った健康上好ましくない出来事に関する情報ということができます。
有害事象(Adverse Event):医薬品が投与された患者あるいは被験者に生じたあらゆる好ましくない医療上の
出来事。
副作用(Adverse Drug Reaction):有害事象のうち当該医薬品との因果関係が否定できない反応。
治験依頼者である製薬会社は、これらの事項を知ったときには、定められた期間内に厚生労働大臣に報告する義務があります。
※製造販売業者は取り扱う医薬品について、厚生労働省令で定める副作用等を知った時は、定められた期間内にその旨を厚生労働大臣に報告しなければならない(2005年3月17日、薬食発0317006号)。
この副作用等の報告の用語については、1999年12月28日付けで、ICH国際医薬用語集日本版(MedDRA/J)も使用できるようになった(1999年12月28日、医薬安.第164号・医薬審1843号)。
また、2003年10月27日以後は電子的副作用報告が求められるようになり、インターネット経由で、あるいは電子媒体と書面を持参または郵送することによって報告することになりました。
|
|
 |
|
|
|
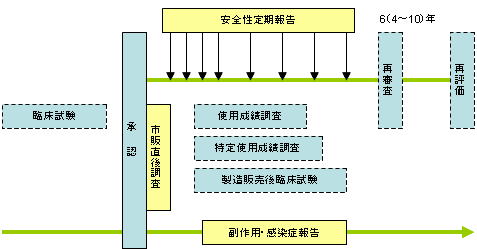
副作用等報告制度の概略 資料:厚生労働省「厚生労働白書」(平成17年版) |
|
 |
|
|
|
市販直後調査 薬事法/GVP
新医薬品の販売開始直後において、医療機関に対し確実な情報提供、注意喚起等を行い、適正使用に関する理解を促すとともに、重篤な副作用及び感染症(以下「副作用等」という。)の情報を迅速に収集し、必要な安全対策を実施し、副作用等の被害を最小限にすることを主な目的とする。新医薬品等の承認の際、承認条件として指示された場合に行う。調査期間は販売開始後六か月間。 |
安全性定期報告
安全性定期報告制度とは、国内で実施される使用成績調査等の結果に加え、世界における安全性情報(PSUR:Periodic Safety Update
Reports)も併せて評価した結果を厚生労働省に報告する制度。承認時に厚生労働大臣が指定した日から2 年間は半年ごと、その後は1 年ごとに厚生労働省に安全性定期報告書を提出する。
|
感染症定期報告
生物由来製品の製造販売業者は、承認日から6ヶ月(厚生労働省が指定する生物由来製品は、指定の期間)ごとに感染症定期報告が必要。(平成15年5月15日医薬発第0515008号) |
副作用・感染症等の報告
製造販売業者は、製品による副作用、感染症等を知ったときは、その重篤度に応じ、15日以内又は30日以内に機構を通じて厚生労働大臣へ届け出なければならない。報告に関する窓口・提出先は総合機構。(薬事法77条4-2、薬事法施工規則64条5-2、平成17年3月17日薬食発第0317006号)
|
定期的安全性最新報告(PSUR:Periodic Safety Update Report)
平成8年11月のICHにおいてPSURのガイドラインが合意された。新薬を開発・販売する各国の製薬メーカーは、関連企業を通じて当該医薬品の副作用の発生動向を
各国の規制当局に提出しなければならない。(薬安第32号、平成9年3月27日) |
治験中の副作用報告
治験中に治験依頼者が入手した副作用等の情報のうち、薬事法施行規則等で定められているものは、治験依頼者が知ったときから7日又は15日以内に厚生省等に報告される。平成15年7月より、いわゆる医師主導の治験の制度が開始されたが、自ら治験を実施する者も同様の報告義務がかかっている。(薬食審査発第0331001号、0331004号、平成17年3月31日) |
製造販売後安全管理業務手順書等の作成 薬事法/GVP
次のような事項を定めること。
・安全管理情報の収集に関する手順
・安全管理情報の検討及びその結果に基づく安全確保措置の立案に関する手順
・安全確保措置の実施に関する手順 ほか
|
製造販売後安全管理に関する業務に従事するものに対する教育訓練 薬事法/GVP
次のような事項を定めること。
・研修計画に関する事項
・教育訓練の対象者及び内容に関する事項
・教育訓練の結果の評価
|
用語
副作用(ADR:Adverse Drug Reaction)
投与された薬物(医薬品含む)に対するあらゆる有害で意図しない反応。
有害事象(AE:Adverse Event)
薬物(医薬品含む)を投与された被験者に生じた あらゆる好ましくない医療上のできごと。必ずしも当該薬物の投与との因果関係が明らかなもののみを示すものではない。
CIOMS(Council for International Organizations of Medical Science):国際医学団体協議会
1949年にWHO(世界保健機構)とユネスコとの協賛により設立されその本部はジュネーブ(スイス)にある。各国の医学関連団体、研究グループ、行政機関がメンバーとなり、国際間にまたがるような医学関連事項の研究推進を行い、国際的な医療関連業務の円滑な促進を図ることを目的としている。
CIOMS−Iは個別症例報告様式の標準化、CIOMS−IIは安全性に関する定期報告様式、システムの標準化といった作業を行っている。CIOMS−IIが基になって、現在のICHのPSURが出来上がっている。
市販医薬品に関する定期的安全性最新報告(PSUR)
ICHにおける三極の合意事項として,その作成のための標準的な方法(原文)がまとめられました。
市販医薬品に関する定期的安全性最新報告(PSUR)について 薬安第32号 平成9年3月27日
新薬を開発・販売する各国の製薬メーカーは、当該医薬品の副作用の発生動向を各国の規制当局に提出します。日本においては、国内における新薬の使用状況にPSURの情報を盛り込んで、「安全性定期報告」として、2年間は半年毎、その後は1年毎に新薬の国内・海外使用状況を厚労省へ報告するよう製薬企業に義務づけられています。
緊急安全性情報
重篤な副作用が発生した時に、医療の現場に必要な副作用情報を迅速かつ的確に伝達する為に、 厚生労働省または製薬会社の判断で緊急的に医師に配布される文書。
|
|
| |
|
|
|
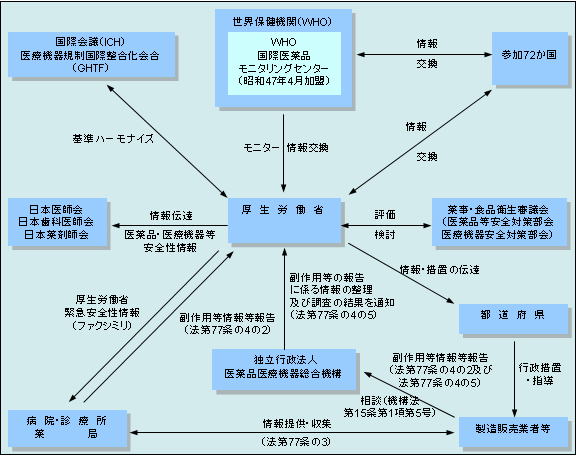
(独)医薬品医療機器総合機構::PMDA |
|
医薬品・医療機器等安全性情報(厚生労働省発行) 独立行政法人 医薬品医療機器総合機構
原則月1回出される医薬品や医療機器の安全性に関する情報
|
副作用が疑われる症例報告に関する情報 独立行政法人 医薬品医療機器総合機構
薬事法によって、製薬会社は副作用と思われる症例があったときは、厚生労働省に報告することが義務づけられています。平成16年4月からは、この報告を独立行政法人
医薬品医療機器総合機構が受けることとなりました。同機構はこれら製薬会社から受けた情報を、平成18年1月31日からすべてラインリスト(新掲載様式)で公開することにしました。これにより、すべての薬品の副作用情報が検索できるデータベースが構築されることになります。
なお、この公表された報告には医療機関等から厚生労働省へ報告された症例も含まれています。
新掲載様式で公開されるのは、平成16年4月分の副作用報告で、その後順次掲載されていきます。
|
緊急安全性情報(ドクターレター) 独立行政法人 医薬品医療機器総合機構
安全性についての緊急かつ重要な情報で、迅速・的確に医療機関に伝達される情報
|
医薬品安全対策通知 独立行政法人 医薬品医療機器総合機構
厚生労働省及び医薬品医療機器総合機構が発出した市販後における医薬品の安全性に関する通知や添付文書の改訂指示通知などの情報を掲載しています。
|
DSU 医薬品安全対策情報 独立行政法人 医薬品医療機器総合機構
医薬品を使う上での新たな注意事項について、製薬業界が取りまとめた情報
|
重篤副作用疾患別対応マニュアル 独立行政法人 医薬品医療機器総合機構
厚生労働省は、薬の重い副作用が原因で起きる疾患の見分け方や治療法などをまとめた「重篤副作用疾患別対応マニュアル(手引書)」を、間質性肺炎など9疾患について作成しホームページで公開しました。(2006年11月)
副作用の早期発見・早期対応の強化を図るため、2005度から4年計画でマニュアルづくりを進めており、最終的には約120の副作用疾患についてマニュアル化を目指しています。
|
医療安全情報 独立行政法人 医薬品医療機器総合機構
医薬品・医療機器に関連する医療安全対策
|
|
|
財団法人日本医療機能評価機構 |
|
|
|
|
|
国立医薬品食品衛生研究所::NIHS |
|
NIHS医薬品安全性情報
- 海外規制機関 医薬品安全性情報 ( 隔週報 )
- 各国公的機関安全性情報
- 医薬品情報ガイド (Drug Info Guide)
-
|
海外公的機関 医薬品安全性情報 国立医薬品食品衛生研究所(NIHS)
海外の主な規制機関、国際機関等から出される医薬品に関わる重要な安全性情報を収集、検討し、日本語の概要等をつけて医薬品安全性情報として迅速に提供することを目的にしています。
|
海外規制機関について 国立医薬品食品衛生研究所(NIHS)
|
|
|
|
|
|
| |
|
|
|
ICH E2B ガイドライン |
|
個別症例安全性報告(ICSR)を伝送するためのデータ項目についてのガイドラインです。 |
|
|
|
|
|
ICH E2C ガイドライン |
|
市販医薬品に関する定期的安全性最新報告(PSUR:Periodic Safety Update Report )についてのガイドラインです。 |
|
|
|
|
|
ICH E2D ガイドライン |
|
「承認後の安全性情報の取扱い:緊急報告のための用語の定義と報告の基準」についてのガイドラインです。 |
|
|
|
|
|
ICH E2E ガイドライン |
|
市販後の医薬品安全性監視についてのガイドラインです。特に新医薬品の市販後早期における安全性監視活動の立案支援が目的で、予測予防対応型の医薬品安全性監視について規定しています。 |
|
|
|
|
|
ICH E2F(開発時定期的安全性最新報告) ガイドライン: |
|
 「ICHトピックE2F Step2ガイドライン説明会」の資料 2008年10月21日、日本製薬工業協会 「ICHトピックE2F Step2ガイドライン説明会」の資料 2008年10月21日、日本製薬工業協会
ICHトピックE2Fは、2008年6月に米国で開催されたICHポートランド会議において、Step 2(ガイドライン案の決定、承認及び各国におけるガイドライン案の内示、意見聴取段階)に到達しました。本ガイドライン案が提案する開発段階における定期的安全性最新報告は、ICH地域に共通する治験安全性年次報告の標準となることを意図して作成されています。
説明会は、本ガイドラインが日本で新しく導入される制度になることから、ガイドライン(案)の内容の説明を中心にして実施されました。これにより、現在実施中のパブコメにおいて、ガイドラインの意図するところを踏まえたコメントを広く募ることが期待されています。
|
ICH E2F:開発時定期的安全性最新報告(案) パブリックコメント募集 2008年9月12日、厚生労働省医薬食品局審査管理課
|
|
| |
|
|
|
日米EU医薬品規制調和国際会議・シカゴ会合 平成18年11月13日
・「臨床安全性データの取扱い(E2B)」の電子的報告の標準作成につき、今後SDOs(Standards Development Organizations)
と共同作業をするための試行を行うことが合意された。
・「医薬品辞書のためのデータ項目及び基準(M5)」については、医薬品辞書に必要なデータ項目及び基準について引き続き検討を行い、2007年のステップ4到達を目指すこととされた。
|
日米EU医薬品規制調和国際会議・横浜会議 平成18年6月15日
・「医薬品規制情報の伝送に関する電子的標準(M2)」及びeCTDについては、eCTDのQ&A(追加)が合意された。
・電子化については今後SDOs(Standards Development Organizations) と共同作業をする方向性が確認された。
・「個別症例安全性報告を伝送するためのデータ項目及びメッセージ仕様(E2B(R3))」については、ステップ3におけるコメント処理の進捗状況が報告され、次回会議でステップ4到達を目指すこととされた。
・「医薬品辞書のためのデータ項目及び基準(M5)」については、メンテナンス手法に関する文書を今後作成することとし、併せて2007年のステップ4到達を目指すこととされた。 |
|
| |
|
|
|
|
| |
|
|
|
|










